
ANOMALII CAUZATE DE ABERATII STRUCTURALE ALE CROMOZOMILOR
Aberatiile structurale ale cromozomilor pot cauza (celor care le poarta sau descendentilor lor), prin diversitatea lor, o gama extrem de variata de efecte nocive. Aceste efecte depind in mare masura de: tipul aberatiei, dimensiunile segmentului cromozomial implicat, importanta genelor implicate si de modul sau masura in care functionarea lor este afectata.
Uneori deletii terminale sau intercalare pot fi rặspunzặtoare de aparitia unor sindroame grave, plurimalformative. Asa este cazul sindroamelor Wolf-Hirschhorn si cri-du-chat care sunt datorate unor deletii la nivelul bratului scurt al cromozomului 4 (4p-), respectiv 5 (5p-). In ambele cazuri se inregistreazặ o intarziere a cresterii si retardare mintalặ gravặ. Denumirea sindromului cri-du-chat provine de la faptul cặ plansul nou-nặscutului seamặnặ (datoritặ dezvoltặrii incomplete a laringelui) cu tipặtul pisicii. Ambele afectiuni sunt foarte rar intalnite, incidenta lor fiind apreciatặ la 1/50.000 de nasteri.
In unele cazuri deletiile cuprind portiuni foarte mici de cromozomi si nu pot fi identificate decat prin tehnici speciale precum bandarea de inaltặ rezolutie in prometafazặ sau FISH. Prin aceste metode au putut fi stabilite cauzele unor sindroame cunoscute in patologia clinicặ dar a cặror origine era necunoscutặ. Aceste sindroame datorate unor microdeletii sunt cauzate de pierderea unui numặr mic de loci adiacenti. Astfel, unii bặieti cu distrofie muscularặ Duchenne (DMD) sunt afectati si de alte maladii determinate de gene X-linkate precum retinita pigmentarặ sau deficienta de glicerol kinazặ. Locii pentru aceste maladii sunt situati foarte aproape de locusul pentru DMD (Xp21).
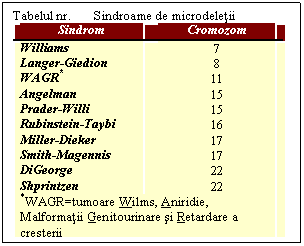
Tabelul nr. prezinta o serie de sindroame produse de microdeletii. Icidenta lor pare mica dar se considera ca aplicarea tehnicilor moderne va permite identificarea cauzelor pentru alte anomalii a caror origine nu este inca cunoscuta. Se apreciaza deci ca frecventa acestor sindroame produse de microdeletii ar putea fi mult mai mare decat se estimeaza in prezent.
 Tumoarea
Wilms
Tumoarea
Wilms
O parte dintre copii care prezinta neoplasm renal embrionar, cunoscut sub numele de tumoare Wilms, au si aniridie (absenta irisului), anomalii genito-urinare si intarzieri in crestere si dezvoltare. Aceasta combinatie de anomalii este cunoscuta sub numele de sindrom WAGR. Analiza cromozomiala la copii afectati a permis identificarea unei deletii interstitiale la nivelul cromozomului 11 (11p13). Studiile moleculare au permis identificarea anumitor gene localizate la nivelul portiunii deletate. Astfel, pierderea genei PAX6 este responsabila de aparitia aniridiei in timp ce lipsa genei WT1 cauzeaza dezvoltarea tumorii Wilms. Aceste date pot fi astazi utilizate in scopul diagnosticarii copiilor cu deletii 11p13 si aprecierii riscului ca acestia sa dezvolte tumoare Wilms. Descoperirile bazate pe studiul sindroamelor de microdeletii sau dovedit a fi deci foarte importante, ele permitand izolarea unor gene ce cauzeaza tumori embrionare.
Sindromul Prader-Willi
In cazul sindromului Prader-Willi, copii sunt puternic hipotoni in primii ani de viata ca ulterior sa devina obezi, au statura mica, hipogonadism si sunt moderat inapoiati mintal. Studiile citogenetice au evidentiat faptul ca 75% din copii afectati acest sindrom prezinta o microdeletie in regiunea proximala a bratului lung al cromozomului 15 (15q11-12). Analiza prin tehnici moderne a evidentiat faptul ca cromozomul afectat este aproape intotdeauna cel provenit de la tata. Restul de 25% de copii afectati nu prezinta microdeletie la nivelul cromozomului 15 ci o disomie uniparentala. Descoperiri recente indica faptul ca doar alela paterna dintr-o anumita regiune a cromozomului 15 (regiune care include gena pentru ribonucleoproteina mica nucleara N-SNRPN) este exprimata, alela materna ca si unele gene adiacente fiind inactive. Aceste constatari sustin ipoteza ca in sindromul Prader-Willi gena pentru SNRPN si unele gene adiacente ei nu se exprima. Aceasta deficienta poate determina, in parte, la simptomele sindromului Prader-Willi.
Sindromul Angelman
Sindromul Angelman se caracterizeaza prin epilepsie, accese anormale de ras, retardare mintala si o slaba capacitate de coordonare a miscarilor (ataxie). Cercetarile au aratat faptul ca 70% dintre cei afectati prezinta o deletie interstitiala a aceleiasi regiuni a cromozomului 15 ca si in cazul sindromului Prader-Willi, doar ca in acest caz cromozomul afectat este de origine materna. 2% din cazurile de sindrom Angelman sunt cauzate de o disomie uniparentala paterna. Se apreciaza ca un mic procent dintre subiectii cu sindromul Prader-Willi si cca. 2-3% dintre cei cu sindrom Angelman isi datoreaza maladia mutatiei la nivelul genelor care controleaza fenomenul de imprinting(imprinting=fenomenul prin care o gena sau o regiune a unui cromozom are o exprimare diferita in functie de originea materna sau paterna). Se apreciaza la restul de 25% din cazuri, pacientii se nasc cu sindrom Angelman datorita unui mozaicism gonadal matern pentru o singura gena mutanta. Recent au fost identificate mutatii ale genei UBE3A, care este una din genele care par a fi preferntial sau exclusiv exprimate de cromozomul 15 provenit de la mama. Se presupune ca mutatiile de la nivelul acestei gene ar determina distrugerea unor proteine la nivelul sistemului nervos central in timpul dezvoltarii. Din cauza mozaicismului gonadal se apreciaza ca, in acest din urma caz, riscul recurentei este semnificativ.
Literatura de specialitate raporteaza cazuri in care sindroamele Angelman si Prader-Willi au fost datorate unei translocatii pentru portiunea proximala a bratului lung a cromozomului 15. In functie de parintele de la care mosteneste translocatia, copilul va prezenta fie sindromul Prader-Willi, fie Angelman.
Sindroamele DiGeorge si Shprintzen
Indivizii cu sindrom DiGeorge apar foarte rar, sub forma unor cazuri sporadice. Cea mai mare parte a indivizilor afectati prezinta malformatii cardiace, tetanii, hipoplazie de timus si paratiroide. Ca urmare a numarului redus sau chiar a lipsei limfocitelor T (ca urmare a hipoplaziei sau lipsei timusului) copii sufera de infectii virale recurente.
Studiile moleculare au aratat ca cea mai mare parte a cazurilor sunt datorate unei microdeletii in regiunea proximala a bratului lung al cromozomului 22 (22q11-2). In mod surprinzator s-a constatat ca indivizii cu sindrom Shprintzen prezinta un defect similar. Acest sindrom se caracterizeaza prin acelasi tip de malformatii cardiace ca si in cazul sindromului DiGeorge, palat despicat si expresie faciala caracteristica. Au fost identificate unele familii in care sindromul Shprintzen pare sa se transmita autosomal dominant. Se apreciaza ca acest sindrom ar fi datorat deletiei unui segment foarte mic de cromozom, motiv pentru care riscul aparitiei unor malformatii grave este mai mic decat in cazul sindromului DiGeorge. Ulterior s-a constatat ca in numeroase familii cu malformatii cardiace, care se credea ca au o determinare multifactoriala, indivizii afectati prezinta microdeletii la nivelul bratului lung al cromozomului 22. Astfel, s-a putut demonstra ca o anomalie care parea sa fie determinata multifactorial, poate fi transmisa dominant autosomal.
SINDROMUL 18q-
Fenotipul persoanelor afectate, desi poate fi foarte variabil, se caracterizeaza in principal prin retadrare mintala, statura mica, hipotonie, deficiente de auz si deformari ale membrelor inferioare. In unele cazuri s-a constatat un nivel scazut al imunoglobulinei A (Hecht, 1969) iar o bolala autimuna ce insoteste deficienta de IgA a fost, de asemenea, intalnita la unii pacienti cu sindrom 18q- (Wilson si colab., 1979). Numeroase studii ( Schwarz si Duck, 1990; Andler si colab., 1992; Ghidoni si colab., 1997) indica faptul ca indivizii afectati au o deficienta de producere a hormonului de crestere (STH) fapt ce conduce la ideea ca una sau unele gene de pe bratul lung al cromozomului 18 sunt implicate in producerea hormonului de crestere. Studiul a 50 de indivizi cu deletii la nivelul 18q a aratat ca aproape toti cei investigati (48 din 50) prezentau greutate scazuta, ritm de crestere redus, valori ale IGF1 si/sau IGFBP3 sub medie sau raspuns deficitar la stimulatori ai STH precum arginina si clonidina (Halle si colab., 2000).
Investigand 33 de copii cu sindromul 18q- si deficienta de hormon de crestere, Cody si colab. (1997) a identificat o regiune de cca. 2 Mb care lipsea la toti cei care prezentau insufienta de STH. Ei au indicat doua gene din aceasta regiune (gena pentru proteina bazica a mielinei- MBP; 159430 si cea pentru receptorul galaninei GLANR; 600377-) ca fiind candidate pentru fenotipul de insuficienta de STH.
Brknac si colab. (1998) a a analizat ADN de la pacienti diagnosticati cu deletii de novo ale portiuni terminale ale cromozomului 18. Ei au realizat analiza moleculara cu markeri polimorfi pe toata lungimea regiunii 18q-.Tehnica FISH a fost efectuata cu doua probe telomerice 18q si cu o proba alfa-satelit specifica cromozomului 18. Din cei 35 de pacienti diagnosticati initial cu deletii terminale 18q, ei au constatat ca 5 (14%) preyentau rearanjamente mai complexe iar 3 (9%) pastrau extremitatea terminala 18q dar aveau o lipsa interstitiala la nivel subterminal. Aceste observatii indica faptul ca realizarea unui cariotip standard este insuficienta pentru caracterizarea sindromului 18q-.
Katz si colab. (1999) a clonat presupusul punct de ruptura (18 q21.3) de la pacientii cu sindrom 18q- Acest punct de ruptura este localizat intre doua gene care codifica inhibitori ai serin-proteazei (SCCA1 si SCCA2). Desi studiile citogenetice sugerau faptul ca aceasta aberasie cromozomiala rezulta ca urmare a unei simple deletii terminale, analiza ADN prin electroforeza in camp pulsatoriu si tehnica FISH au aratat faptul ca punctul de ruptura este in vecinatatea unei secvente de 35 pb urmata de o secventa de ADN satelit III ce contine un fragment telomeric de 475-1000kb.Acest tip de secvente de ADN satelit III nu a fost detectat la cromozomul 18 normal dar prezenta un grad inalt de omologie cu secventele de ADN satelit III localizate la nivelul bratelor scurte ale cromozomilor acrocentrici. Aceste observatii sugereaza faptul ca aberatia cromozomiala de tip 18q- survine in urma unei recombinari intre cromozomi neomologi.
|
Politica de confidentialitate |
| Copyright ©
2025 - Toate drepturile rezervate. Toate documentele au caracter informativ cu scop educational. |
Personaje din literatura |
| Baltagul caracterizarea personajelor |
| Caracterizare Alexandru Lapusneanul |
| Caracterizarea lui Gavilescu |
| Caracterizarea personajelor negative din basmul |
Tehnica si mecanica |
| Cuplaje - definitii. notatii. exemple. repere istorice. |
| Actionare macara |
| Reprezentarea si cotarea filetelor |
Geografie |
| Turismul pe terra |
| Vulcanii Și mediul |
| Padurile pe terra si industrializarea lemnului |
| Termeni si conditii |
| Contact |
| Creeaza si tu |